Amazon Genomics CLIでのプロジェクト作成と実行
この章ではnf-coreで公開されているRNAseqのパイプラインを、Amazon Genomics CLIで実行します。
以下のコマンドで新しくAmazon Genomics CLIのプロジェクトを作成します。
cd ~/environment
mkdir -p rnaseq
cd rnaseq/
agc project init RNASeq --workflow-type nextflow
上記コマンドが完了すると~/environment/rnaseq配下にagc-project.yamlが作成されます。ファイルブラウザにてダブルクリックしてファイルを開き、以下の内容を貼り付けます。
name: RNASeq
schemaVersion: 1
workflows:
rnaseq;
type:
language: nextflow
version: 1.0
sourceURL: ./
data;
- location: s3://1000genomes
readOnly: true
- location: s3://nf-core-awsmegatests
readOnly: true
- location: s3://ngi-igenomes
readOnly: true
contexts:
ctx1:
engines:
- type: nextflow
engine: nextflow
上記の設定では、rnaseqという名前のワークフローを作成し、プロジェクトで利用するS3バケットをdataにて読み込み専用readOnly: trueにて設定しています。また、Contextで利用するワークフローエンジンとしてNextflowを指定しています。
MANIFEST.jsonを作成します。
cd ~/environment/rnaseq
touch MANIFEST.json
作成したMANIFEST.JSONに以下の内容を貼り付けます。
{
"mainWorkflowURL": "https://github.com/nf-core/rnaseq.git",
"inputFileURLs": [
"inputs.json"
],
"engineOptions": "-r 3.4 -resume -profile test"
}
inputs.jsonを作成します。
cd ~/environment/rnaseq
touch inputs.json
作成したinputs.jsonに以下の内容を貼り付けます。
{
"input": "https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/samplesheet/v3.4/samplesheet_full.csv",
"genome": "GRCh37"
}
以下のコマンドでContextをデプロイします。
agc context deploy ctx1
以下のコマンドでワークフローを実行します。
agc workflow run rnaseq -c ctx1
実行の進捗は以下で確認できます。
agc workflow status
前章までと同様に、AWS Batchのコンソールから確認することもできます。
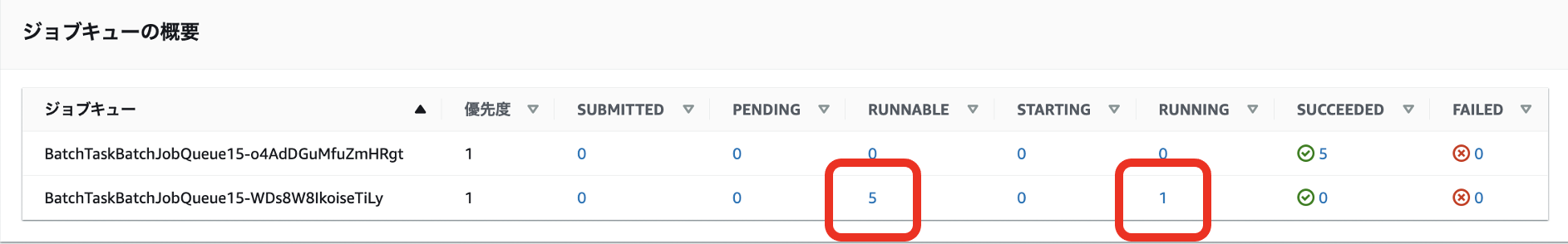
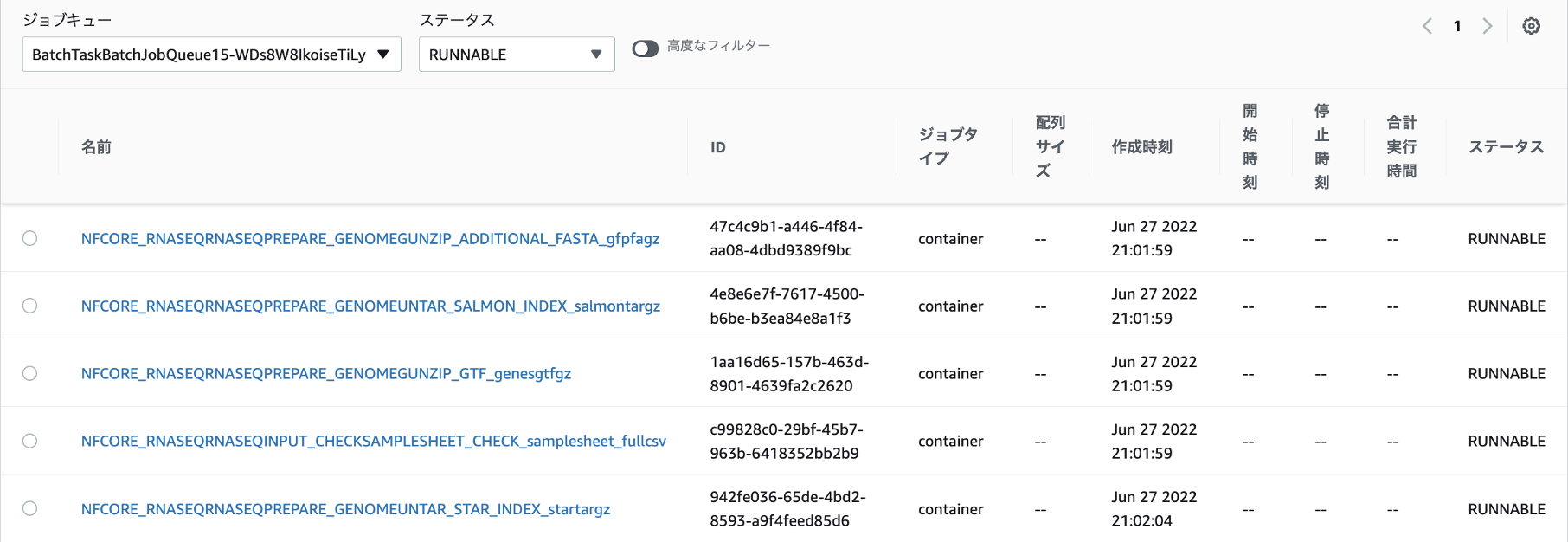
nf-coreのパイプラインで指定されているBiological Replicateなどのデータが並列にジョブ実行されていることが確認できます。
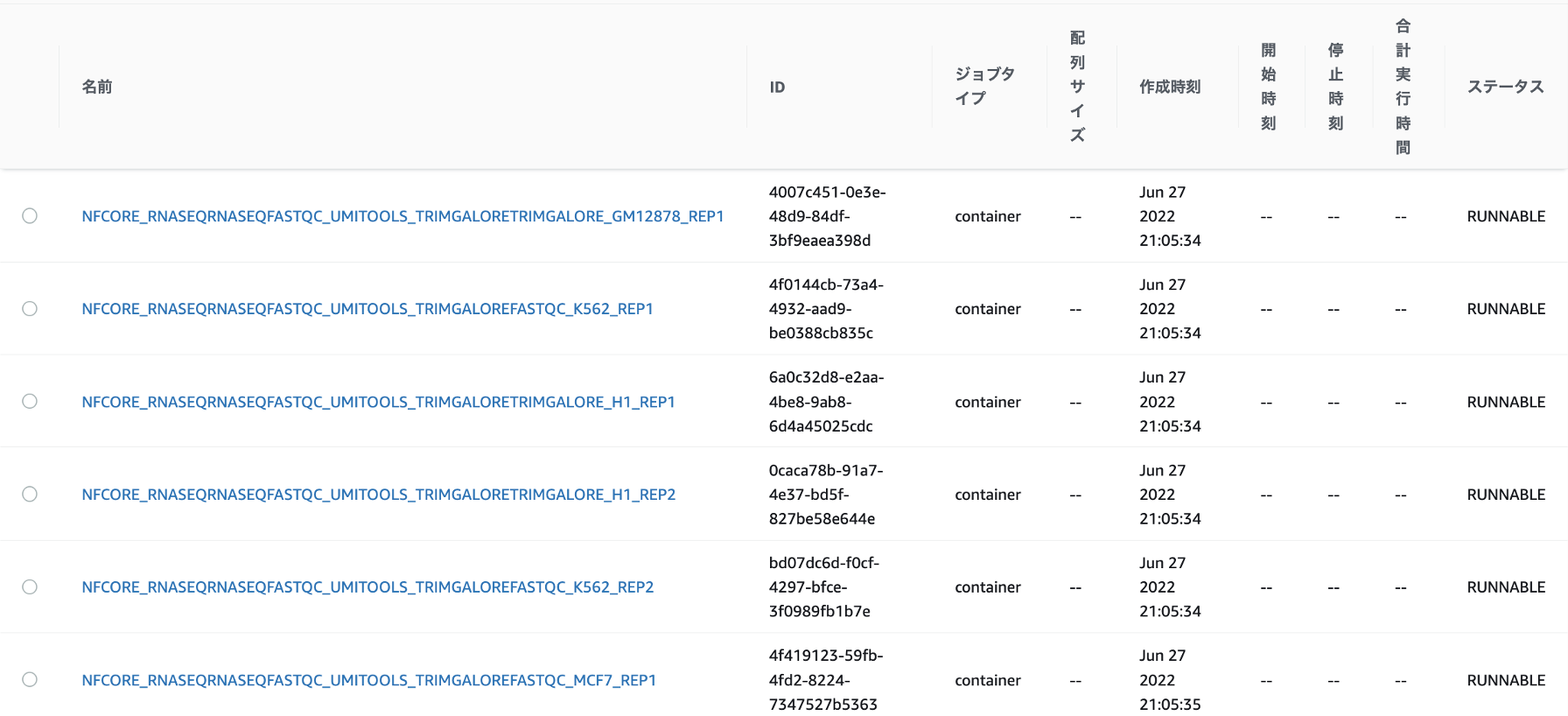
以上でnf-coreのRNAseqパイプラインの実行およびジョブの進捗状況の確認手順の解説を行いました。ハンズオンの中ではこのジョブは完了しませんので、時間をおいてから、ハンズオン環境提供期間内に、再度ジョブの進行状況を確認してみてください。
お疲れ様でした!これにてハンズオンコンテンツは完了です!