簡易RNAseqワークフローの実行
簡易的なRNAseqワークフローを実装/実行する
本章では、ゲノム解析の中でも特にRNAseqについてのワークフローの一部のNextflowでの実装例と実行を通して、ご自身でNextflowを利用したワークフローを実装し、Amazon Genomics CLIを利用してAWS上で実行することで、手軽にクラウドのスケールメリットを享受した解析ができることを確認します。
本章で実行する簡易的なRNAseqワークフローでは以下のオープンソースソフトウェアを利用します。
- FastQC (クオリティチェック)
- Trimmomatic (アダプター配列除去)
- STAR (index作成・マッピング)
以下のようなワークフローを利用します。(なお、生物学的な解釈はこのハンズオンでは対象外とします)
S3バケットへのデータの転送
Cloud9環境に戻り、以下のコマンドを実行して、先程バックグラウンドでダウンロードしておいたデータをS3バケットに転送します。
cd ~/environment
AGC_BUCKET=$(aws ssm get-parameter --name /agc/_common/bucket --query 'Parameter.Value' --output text --region ap-northeast-1)
aws s3 cp ./data s3://${AGC_BUCKET}/data --recursive
利用するワークフローのコードをアップロードします。まず、アップロード先のディレクトリを作成します。
cd ~/environment
mkdir selfrnaseqworkflow
cd selfrnaseqworkflow
以下の手順に従い、アップロードを行います。
selfrnaseqworkflowをファイルエクスプローラー上で選択した状態にし、Upload Local Filesをクリックします。
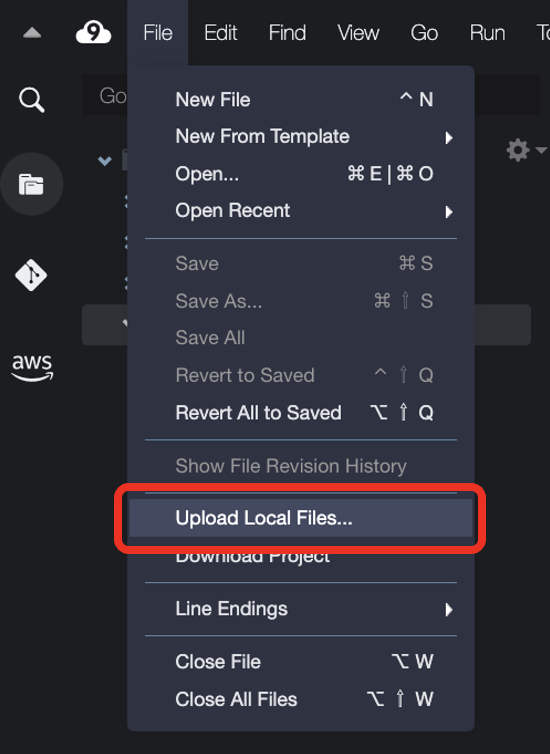
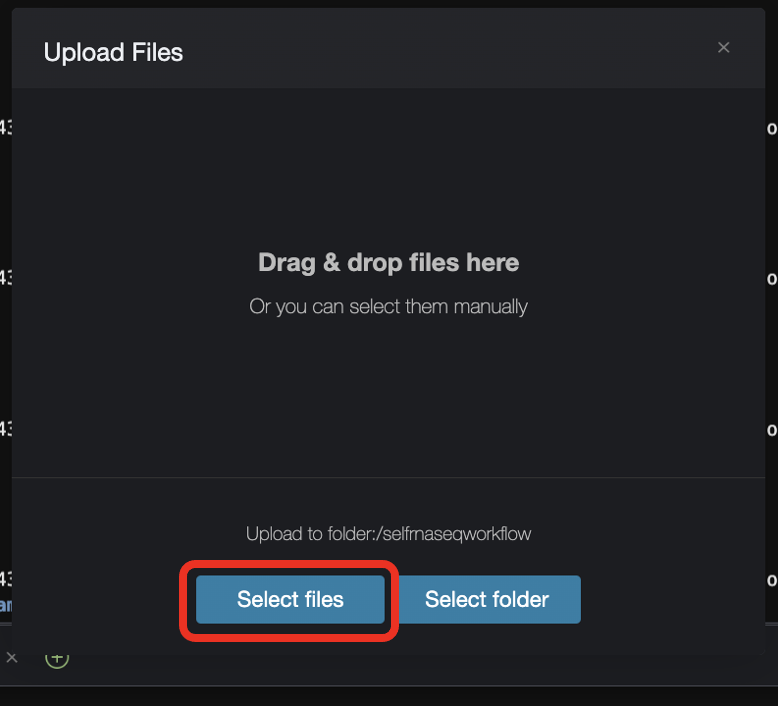
Select fileでダウンロードしたファイルを選択し、アップロードします。アップロード対象は以下の5つのファイルです。
agc-project.yamlinputs.jsonmain.nfMANIFEST.jsonnextflow.config
ワークフローコードの修正
Cloud9上でmain.nfおよびinput.jsonファイル記載のS3バケット名を変更します。まず、変更後のバケット名を以下のコマンドで出力します。
echo $AGC_BUCKET
以下の手順で修正を行ってください。
- ファイルエクスプローラーにて
main.nfとinputs.jsonをダブルクリックし、開きます。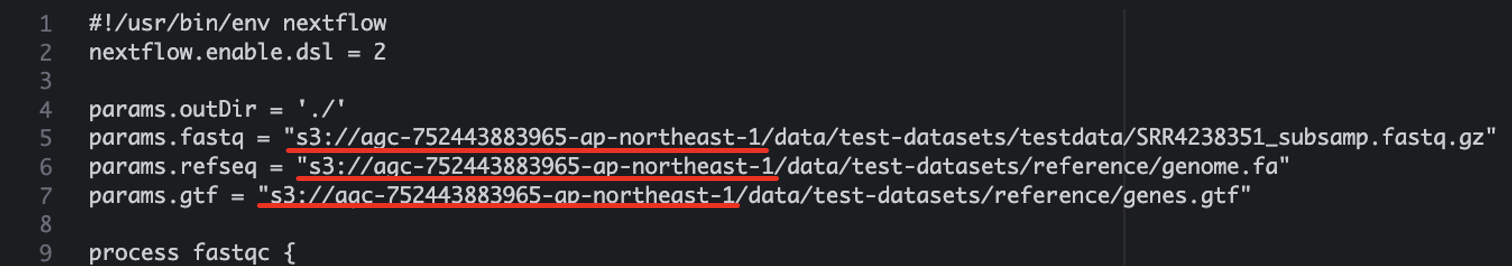
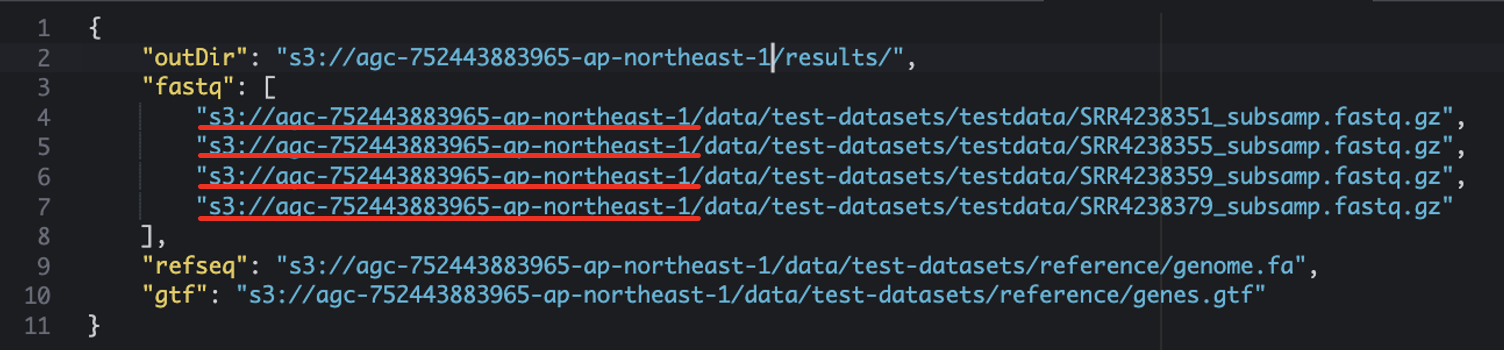
Ctrl + F (command + F)で検索バーを開きます。agc-752443883965-ap-northeast-1を検索し、右側の置換先として、上の手順で取得したAGC_BUCKETの値を入力します。
Replace Allで全て置換します。
ワークフローの実行
新たなProjectとして簡易RNAseqワークフローを実行します。まずは、Contextのデプロイから行います。以下コマンドを実行してください。
cd ~/environment/selfrnaseqworkflow
agc context deploy ctx1
実行の完了を待つ間に、簡易RNAseqをNextflowでどのように記述したのかをみてみましょう。
まずは、~/environment/selfrnaseqworkflow配下の、main.nfを見てみます。main.nfでは、パラメータが利用できます。このパラメータはワークフロー実行時にコマンドラインから上書きすることも可能となっています。
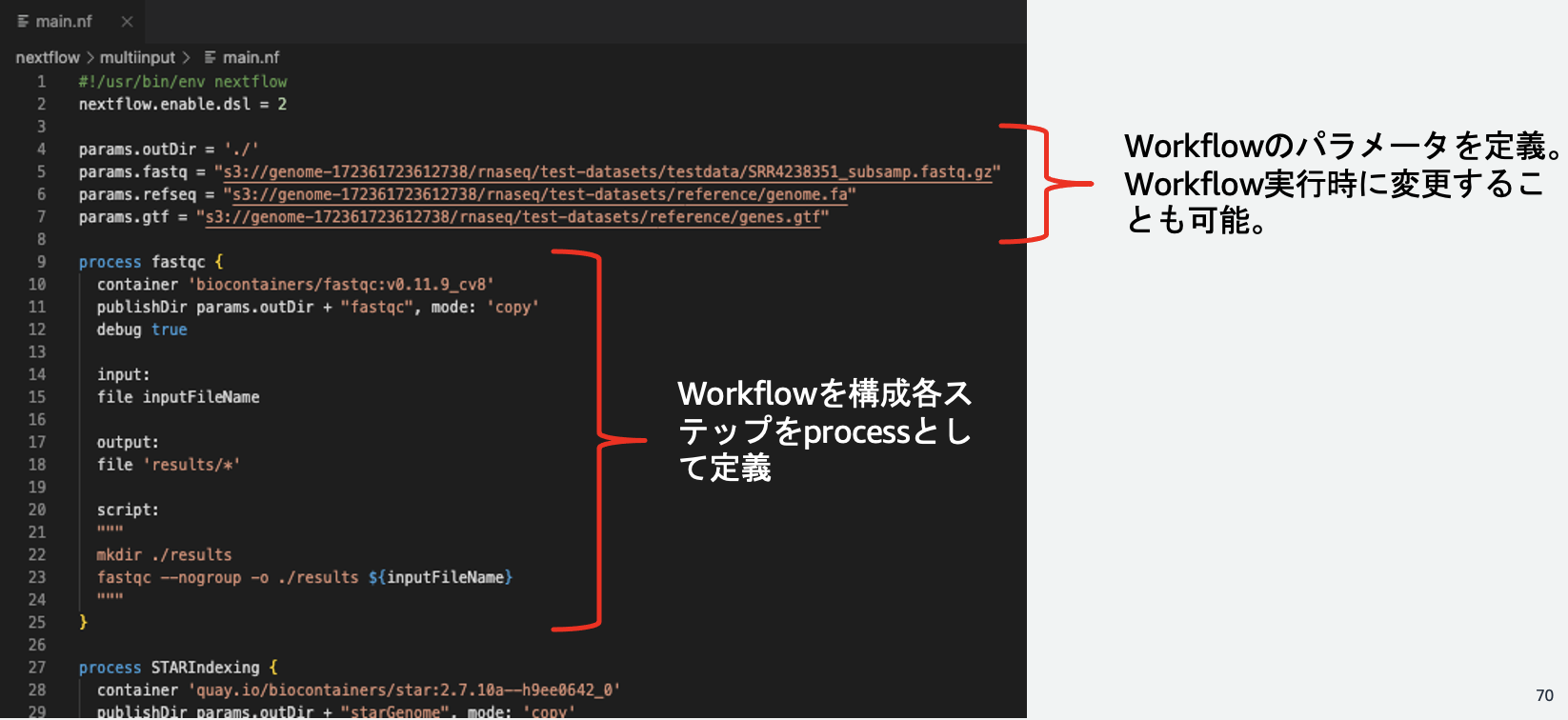
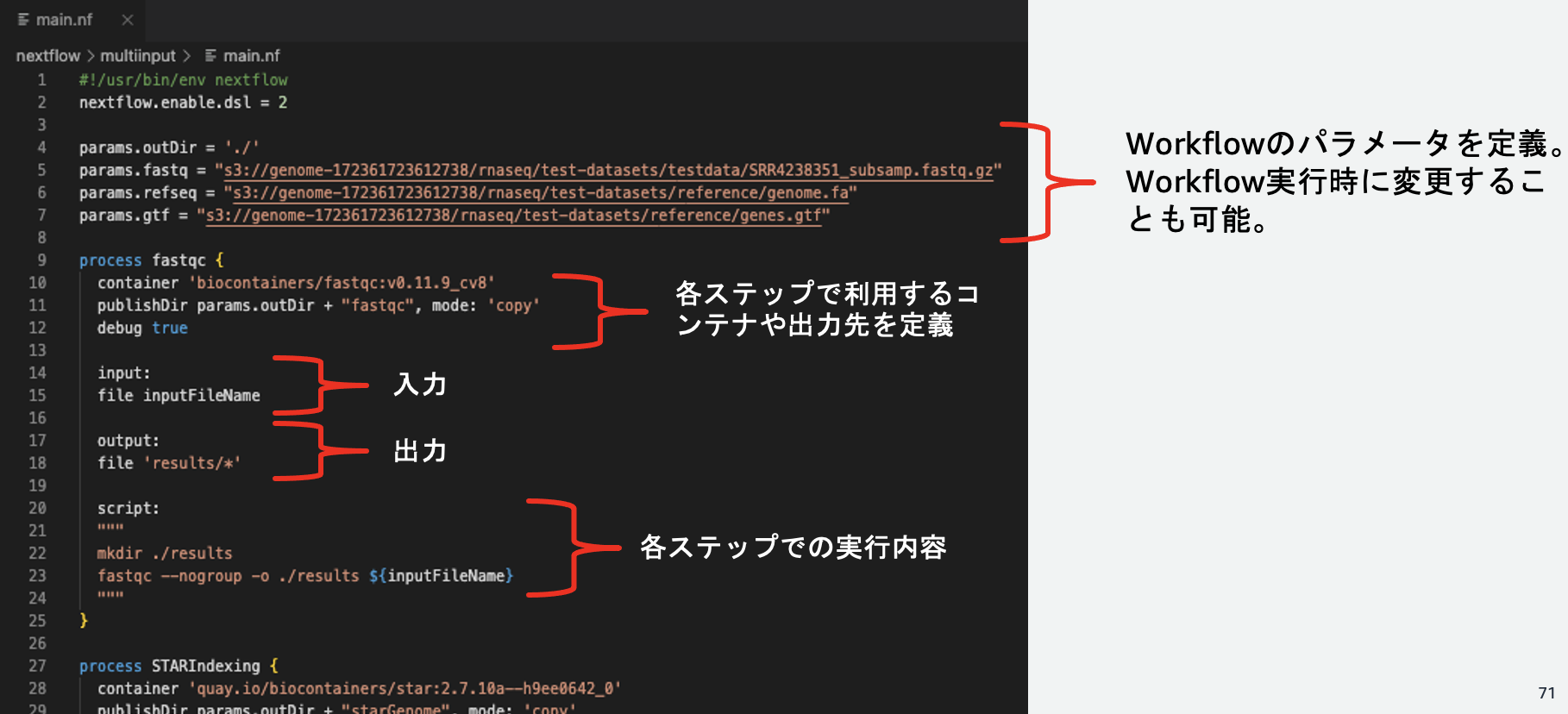
また、helloワークフローでも登場したprocessは先ほどよりも設定項目が増えていることがわかります。process fastqcを見てみましょう。
上から順に設定項目を見ていくと、まずは、containerによりprocessが実行されるコンテナ環境を指定しています。このように、ワークフローhelloではnextflow.configにて実行コンテナ環境設定を行なっていましたが、main.nfに記述した個々のprocessの定義の中で指定することもできます。今回は、biocontainerで公開されているFastQCのコンテナイメージを利用しています。
また、publishDirとoutputにて、出力の設定を行っています。
同様に、TrimmomaticやSTARを実行するprocessもみてみましょう。各ブロックが、入力・出力・実行するコマンド・実行環境などを指定することで構成されていることがわかります。
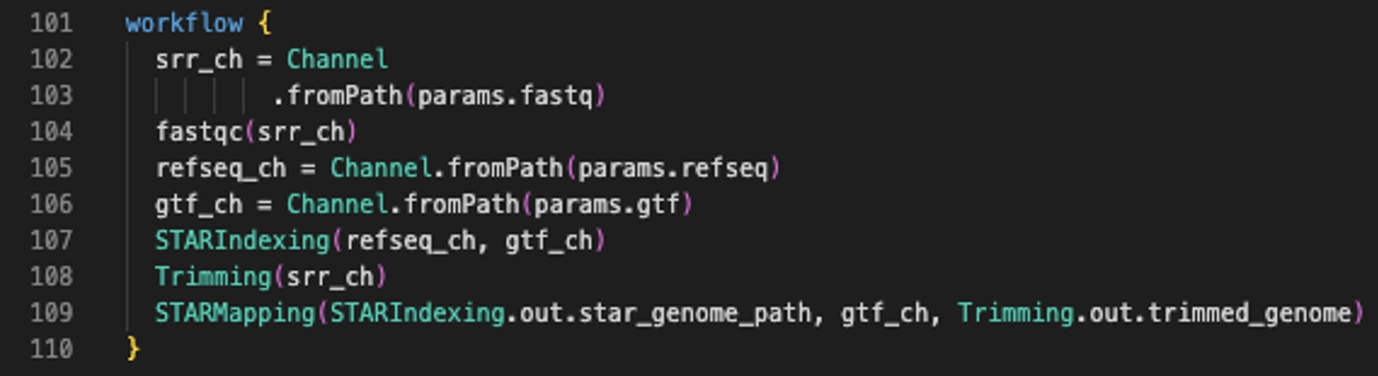
さらに、上で解説した個々のprocessをワークフローとして実行するためには、複数のprocessをつなぐことが必要です。この接続をworkflowにて記述します。
以下のように、各processはその入出力の受け渡しによりワークフローを構築します。前のprocessの出力を入力としたprocessは、前のprocessの完了を待ってから処理を開始します。
そのほかにも様々なワークフローの記述方法やファイル分割による開発効率の向上の方法などがあります。詳しくは公式ドキュメントを参照してください。
さて、今回利用するContextのデプロイが完了したはずです。以下のコマンドでワークフローを実行してみましょう。
agc workflow run rnaseq -c ctx1
本章では、Amazon Genomics CLIでの出力確認は省略します。適宜、前章の内容を元にAmazon Genomics CLIでの結果確認を行ってみてください。ここでは、AWS Batchコンソールから、先ほどと同様にジョブの実行状況を確認してみましょう。
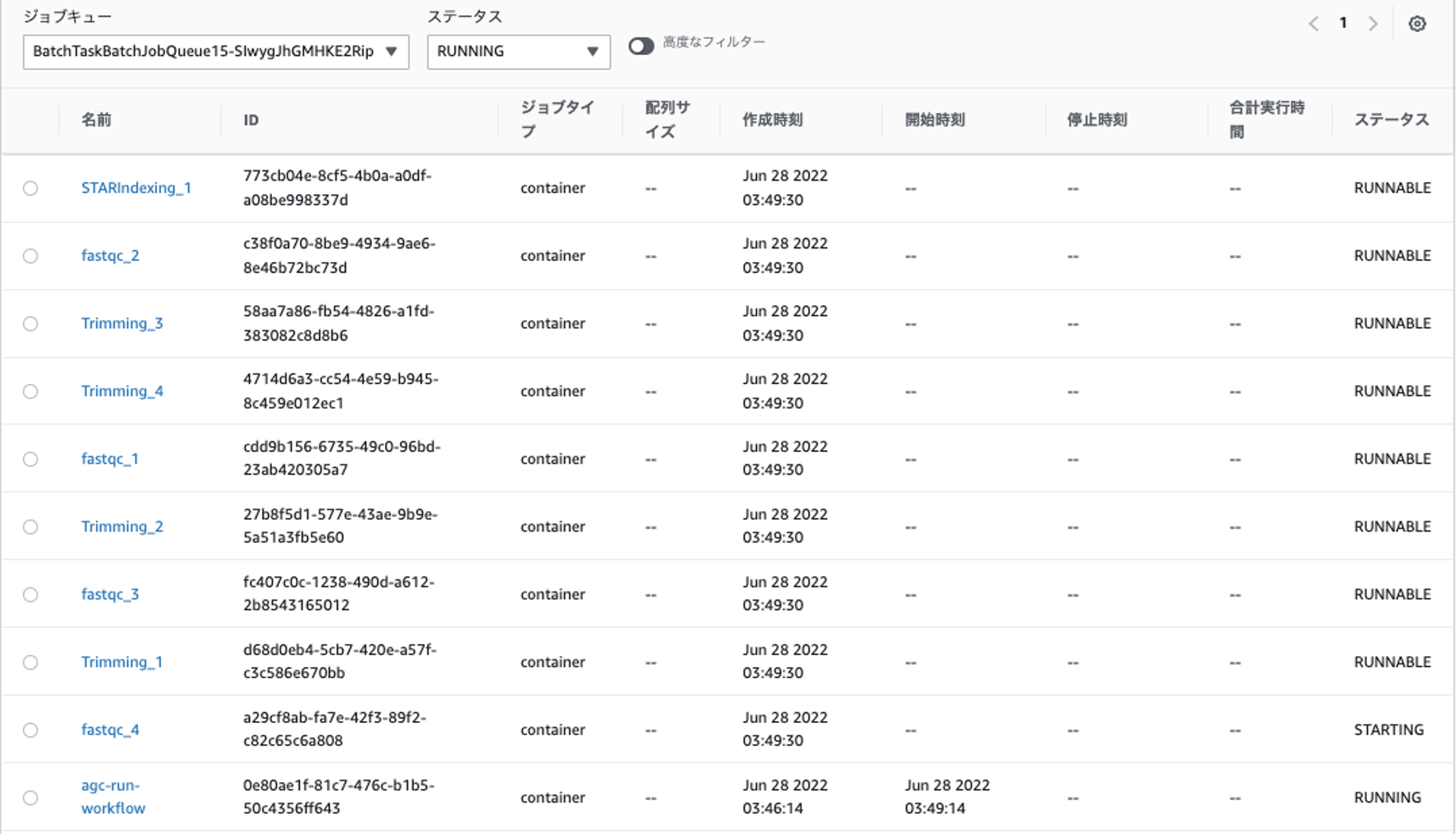
各processが並列にスケールして別々のジョブとして実行されていることが確認できました。また、inputs.jsonにはワークフローに投入する4つの配列を記述しています。このinputs.jsonを編集していただき、入力データを増やすことで、手軽に、さらに多くのジョブを並列投入することが可能となります。
ここまでで、簡易RNAseqワークフローの実行手順は終了です。次に、これらの解析結果をローカル環境にダウンロードすることなしに、クラウド上で可視化してみましょう。